
New research from the RIKEN Center for Brain Science (CBS) in Japan shows that a deficit in histone methylation could lead to the development of autism spectrum disorders (ASD). A human variant of the SUV39H2 gene led researchers to examine its absence in mice. Published in Molecular Psychiatry, the study found that when absent, adult mice exhibited cognitive inflexibility similar to what occurs in autism, and embryonic mice showed misregulated expression of genes related to brain development. These findings represent the first direct link between the SUV39H2 gene and ASD.
Genes are turned on and off throughout our development. But genetic variation means that what is turned off in some people remains turned on in others. This is why, for example, some adults can digest dairy products and others are lactose intolerant; the gene for making the enzyme lactase is turned off when some people become adults, but not others. One way that genes can be turned on and off is through a process called histone methylation in which special enzymes transfer methyl groups to histone proteins that are wrapped around DNA.
Variations in genes related to methylation during brain development can lead to serious problems. One such variation occurs in a rare disorder called Kleefstra syndrome, in which a mutation prevents methylation of H3K9—a specific location on histone H3. Because Kleefstra syndrome resembles autism in some ways, RIKEN CBS researchers led by Takeo Yoshikawa looked for autism-specific variations in genes that can modify H3K9. Among nine such genes, they found one variant in an H3K9 methyltransferase gene—SUV39H2—that was present in autism, and the mutated SUV39H2 prevented methylation when tested in the lab. Similar loss-of-function results were found for the mouse version of the variant.
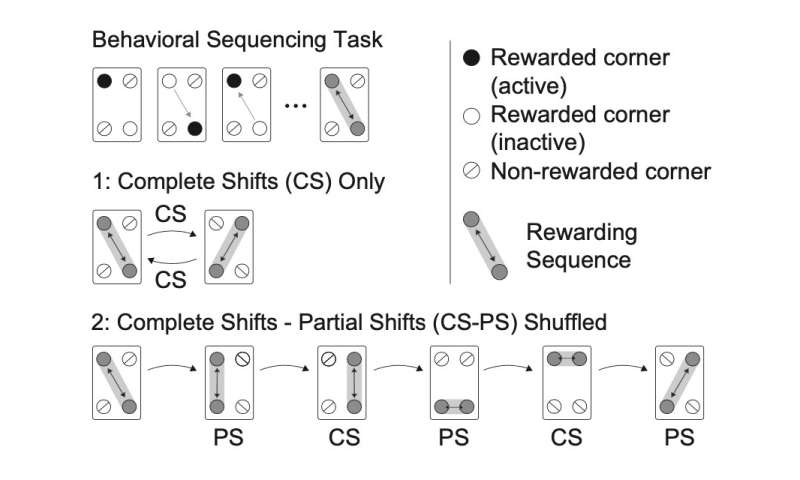
The next step was to see what happens in mice that lack the Suv39h2 gene. Behaviorally, the researchers found that the mice could learn a simple cognitive task, but had difficultly when the task required cognitive flexibility. In the simple task, mice learned to get a reward by poking a door at alternating diagonal corners of a cage. After they could do this well, the possible reward locations switched to the other two diagonal corners. The genetically modified mice did this as well as wild-type mice. In another task, after learning to alternate between the two diagonal corners, only the location of one reward was switched. When the mice were challenged to alternate randomly between these two tasks, wild-type mice could adapt quickly, but the Suv39h2-deficient mice took much longer. “This serial reversal-learning task was essential,” says first author Shabeesh Balan. “Cognitive inflexibility is a core symptom of ASD, and our new task was able to address this behavioral feature in ways that previous mouse studies could not.”
When the researchers examined what happened in the mouse brain when H3K9 methylation failed to occur, they found that important genes that are usually silenced in early development were turned on in the experimental mice. “Suv39h2 is known to be expressed in early neurodevelopment and to methylate H3K9,” explains Yoshikawa. “This keeps a check on genes that should be switched-off. But without it, genes in the protocadherin β cluster were abnormally expressed at high levels in embryonic mice.” Because protocadherins are critical for the formation of neural circuits, the researchers believe they have found an important biological pathway that could be central to several neurodevelopmental disorders.
The team then verified the importance of SUV39H2 in human ASD by finding that its expression was lower in the postmortem brains of people with ASD than of controls. “What began with a loss-of-function mutation in only one person with ASD,” says Yoshikawa, “has led to a general causal landscape for ASD that culminates in brain circuit abnormality.”
Source: Read Full Article